
Die senetics healthcare group GmbH & Co. KG betreibt ein zertifiziertes Prüflabor mit langjähriger Erfahrung bei der Testung von Medizinprodukten nach EN 60601.
Wir prüfen für Sie:
- Die elektrische Sicherheit medizinisch elektrischer Geräte.
- Die funktionale Sicherheit nach DIN EN 60601-1.
- Die Gebrauchstauglichkeit medizinisch elektrischer Geräte gemäß DIN EN 60601-1-6.
- Alarmsysteme in medizinisch elektrischen Geräten gemäß DIN EN 60601-1-8.
- Die Anforderungen an ME-Systeme und Geräte für medizinische Versorgung in häuslicher Umgebung gemäß DIN EN 60601-1-11.
- Die Einhaltung der IEC 62304.
- Die Biokompatibilität nach DIN EN ISO 10993
Die Erfüllung der Basisnorm EN 60601-1 erfordert eine umfassende Prüfung des Medizinproduktes mit Hilfe verschiedener Testungen. Die senetics healthcare GmbH & Co. KG berät Sie bei der Auswahl der nötigen Testungen und bietet die Prüfung wichtiger Leistungsmerkmale an, die im folgenden beschrieben werden.
Elektrische Medizinprodukte in feuchter Umgebung – Feuchtevorbehandlung nach EN 60601-1
Um das Auftreten von Ableit- und Berührströmen beim Arbeiten in feuchter Umgebung auszuschließen, wird am Medizinprodukt ein Funktionstest nach Feuchtevorbehandlung durchgeführt.
Für die Feuchtevorbehandlung wird das Medizinprodukt im Idealfall vollständig in einer Klimakammer aufgebaut. Die Klimakammer wird auf eine relative Feuchte von 93% und eine Temperatur zwischen 20°C und 30°C eingestellt. Prüflinge mit einem Gehäuse der Schutzklasse IPX0 verbleiben für 2 Tage, Prüflinge mit erhöhtem Schutz gegen das Eindringen von Flüssigkeiten für 7 Tage in der Feuchtekammer. Nach der Inkubation wird das Gehäuse auf die Ansammlung von Flüssigkeiten geprüft, welche zu gefährlichen Kriechströmen führen könnten.
Zum Ausschluss einer Gefährdung des Patienten oder Anwenders durch Flüssigkeitsansammlung im Gerät werden folgende Parameter gemäß EN 60601-1 überprüft:
- Erdableitstrom
- Berührungsstrom
- Patientenableitstrom
- Patientenhilfsstrom
- Spannungsfestigkeit
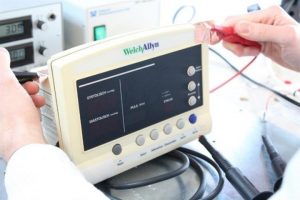
Abbildung 1: 2021_03_11_senetics_Zur Messung des Patientenableitstroms wird eine Metallfolie auf der Oberfläche des Geräts befestigt. Anschließend werden auftretende Ströme vermessen.
Anwendungsteile und berührbare Teile – Unterschiede und Notwendigkeit bei Medizinprodukten
Als „berührbare Teile“ gelten alle Teile des medizinisch-elektrischen Gerätes, welche durch einen Normprüffinger berührt werden können und keine Anwendungsteile darstellen. „Anwendungsteile“ sind ausschließlich die Teile des ME-Geräts, welche bei bestimmungsgemäßem Gebrauch zwangsläufig in physischen Kontakt mit dem Patienten kommen. Die Bestimmung dieser Teile muss grundsätzlich vor Beginn der Testreihe erfolgen, da diese Begriffe im Laufe der Normprüfung fortwährend zur Anwendung kommen und dafür vorab klar definiert werden müssen.
Um zu verhindern, dass Personen mit gefährlichen mechanischen oder elektrischen Teilen des Medizinproduktes in Berührung kommen, werden alle potentiell gefährlichen Stellen am Gerät mit einem Prüffinger oder Prüfhaken analysiert. Der Prüffinger ist kleiner als ein durchschnittlicher menschlicher Finger, um zu gewährleisten, dass Patient oder Anwender nicht ins Innere des Geräts greifen können. Für Geräte, die potentiell in Kontakt mit Kindern kommen, gibt es spezielle Prüffinger in Kindergröße. Mithilfe des Prüffingers werden Kontakt- und Durchgangsprüfungen vorgenommen.

Abbildung 2: 2021_03_11_senetics_Mithilfe eines Prüffingers werden die berührbaren Teile überprüft. Diese Testung dient der Überprüfung der Erreichbarkeit potentiell gefährlicher Stellen.
Die Begleitpapiere eines Medizinprodukts schließen eine Gebrauchsanweisung und eine technische Beschreibung ein. Diese gelten als fester Bestandteil eines jeden medizinischen-elektrischen Geräts und können als elektronische Datei oder in Papierform beigefügt werden. Die Erstellung normenkonformer Begleitpapiere stellt einen wichtigen Beitrag zur Vermeidung unsachgemäßer Benutzung und zur Wahrung der Sicherheit für den Anwender und den Patienten dar.
Sicherheit durch Brandverhütung elektrischer Medizinprodukte
Auch die Beachtung der Brandverhütung bei der Entwicklung von elektrischen Medizinprodukten ist in der EN 60601-1 festgehalten. Hier ist festgelegt, dass das Gehäuse die erforderliche Festigkeit und Robustheit aufweisen muss, um einen durch vernünftigerweise vorhersehbaren Missbrauch oder selten auftretende funktionale Fehler ausgelösten Brand zu vermeiden.
Werden Medizinprodukte in mit Sauerstoff angereicherten Umgebungen betrieben, oder arbeiten sie selbst mit Sauerstoff, gilt es besondere Schutzmaßnahmen zu ergreifen. Aufgrund der brandfördernden Wirkung von Sauerstoff können auftretende Defekte am Medizinprodukt hier zu äußerst gefährlichen und nicht mehr beherrschbaren Komplikationen führen. Die zur Ausbreitung eines möglichen Brandes getroffenen Maßnahmen müssen geprüft werden. Wird das Brandrisiko danach als unvertretbar angesehen, müssen weitere Schutzmaßnahmen ergriffen oder reDesigns am Produkt durchgeführt werden, bis das getestete Restrisiko akzeptabel ist.
Auswahl und Bestimmung sicherer Materialien – Biokompatibilität
Biokompatibilität beschreibt die Verträglichkeit von Werkstoffen mit natürlichem Gewebe. Ein in den Körper eingebrachter Werkstoff (z.B. im Zuge eines Implantates) oder ein mit dem Körper in Kontakt kommendes Gerät darf keinen negativen Einfluss auf das umgebende Gewebe ausüben.
Viele medizinisch-elektrische Geräte kommen in Kontakt mit biologischem Gewebe, Zellen oder Körperflüssigkeiten. Hier verweist die Norm DIN EN 60601-1 auf die Normreihe DIN EN ISO 10993, nach welcher Geräte und Implantate auf ihre biologische Verträglichkeit bewertet und getestet werden. Dabei wird nach der Zweckbestimmung, Verweildauer und Invasivität des Medizinproduktes unterschieden. Die Norm beschreibt unter anderem folgende Prüfungen auf Biokompatibilität:
- Genotoxizität, Karzinogenität, Reproduktionstoxizität (nach DIN EN ISO 10993-3)
- Hämokompatibilität (nach DIN EN ISO 10993-4)
- Zytotoxizität(nach DIN EN ISO 10993-5)
- Pyrogenität/Endotoxine (nach DIN EN ISO 10993-11)
Die funktionale Sicherheit eines Medizinproduktes ist ausschließlich gewährleistet, wenn die Biokompatibilität des Medizinproduktes durch ausreichende Testungen bestätigt ist.
Zusammenfassung
Die zunehmende Komplexität von Medizinprodukten spiegelt sich in einer wachsenden Regulierung des Weges zur Zulassung wider. Diese Regulierung dient der Risikominimierung und der Gewährleistung einer zuverlässigen und sicheren Funktionalität des Medizinprodukts und nützt so dem Anwender wie auch dem Hersteller. Die Normenkonformität sollte schon in frühen Phasen des Entwicklungsprozesses berücksichtigt werden, um auftretende Kosten durch nachträgliche Anpassungen zu vermeiden.
Gerne unterstützen wir von senetics Sie von Anfang an bei der Entwicklung und Prüfung Ihres Medizinproduktes nach DIN EN 60601-1.